The RNA atlas: a nucleotide resolution map of the human transcriptome
Abstract
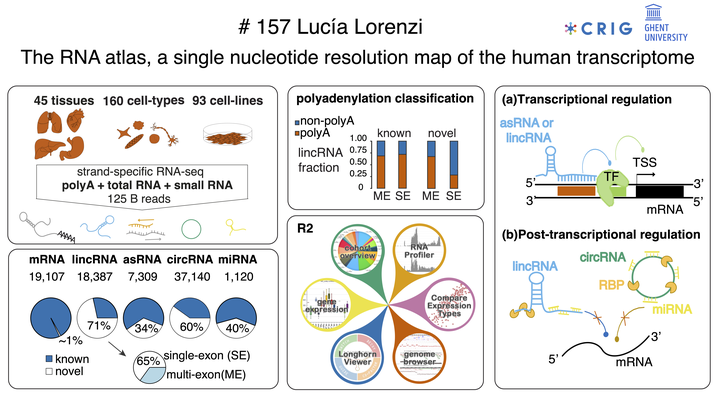
The introduction of RNA-sequencing has enabled us to interrogate the human transcriptome at nucleotide resolution, revealing distinct RNA biotypes beyond protein-coding RNAs. Several consortium-based efforts have contributed to the discovery and quantification of these RNA biotypes in heterogeneous sample collections. However, these studies have mostly applied RNA-sequencing technologies dedicated to the small RNA and polyadenylated RNA fraction of the transcriptome. As a result, a systematic survey of the non-polyadenylated and circularized transcriptome is currently missing. To capture the full diversity of the human transcriptome, we applied complementary RNA-seq methods on a heterogenous collection of 300 human samples including 45 tissues, 162 cell types and 93 cell lines. From these samples, strand-specific polyA, total RNA and small RNA libraries were generated and deeply sequenced to a total of 125 billion reads. We assembled transcripts representing 5 major RNA biotypes (mRNAs, lincRNAs, asRNAs, circRNAs and miRNAs) resulting in 50 235 stringently selected gene loci, of which 19 668 are novel. We identified a handful of novel protein-coding genes with supporting peptides from mass spectrometry data. The majority of novel loci were non-coding in nature and highly enriched for non-polyadenylated single-exon lincRNAs. We provide evidence that these do not derive from contaminating DNA and show that their expression profiles are correlated to underlying sample ontology relationships. We leveraged the broad intron coverage from the total RNA-sequencing data to reveal non-coding RNA regulatory interactions. These analyses revealed that lincRNAs mainly operate at the transcriptional level while circRNA function is mostly post-transcriptional. When applying this concept to the novel miRNAs, 347 miRNAs showed evidence for post-transcriptional regulation of their predicted target genes. Taken together, the RNA-Atlas dataset complements, and extends beyond the scope of other human expression atlases. The dataset serves as a community resource to mine the expression landscape of various RNA biotypes, including a unique collection of non-coding RNAs, and their regulatory interactions. Data and results will be made available through the R2 Genomics Analysis and Visualization Platform.