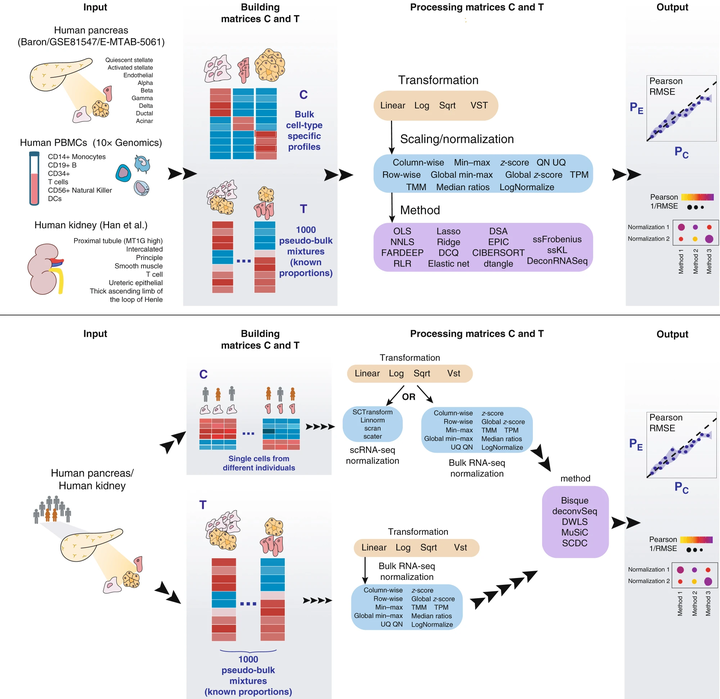
 Schematic representation of the benchmarking study
Schematic representation of the benchmarking study
Abstract
Many computational methods have been developed to infer cell type proportions from bulk transcriptomics data. However, an evaluation of the impact of data transformation, pre-processing, marker selection, cell type composition and choice of methodology on the deconvolution results is still lacking. Using five single-cell RNA-sequencing (scRNA-seq) datasets, we generate pseudo-bulk mixtures to evaluate the combined impact of these factors. Both bulk deconvolution methodologies and those that use scRNA-seq data as reference perform best when applied to data in linear scale and the choice of normalization has a dramatic impact on some, but not all methods. Overall, methods that use scRNA-seq data have comparable performance to the best performing bulk methods whereas semi-supervised approaches show higher error values. Moreover, failure to include cell types in the reference that are present in a mixture leads to substantially worse results, regardless of the previous choices. Altogether, we evaluate the combined impact of factors affecting the deconvolution task across different datasets and propose general guidelines to maximize its performance.