The lab of Prof. Jo Vandesompele and Prof. Pieter Mestdagh at Ghent University
The OncoRNALab is supervised by Prof. Jo Vandesompele and Prof. Pieter Mestdagh at Ghent University,
Department of Biomolecular Medicine,
Faculty of Medicine and Health Sciences. It is part of the
Center for Medical Genetics and the
Cancer Research Institute Ghent.
The lab’s research aims to exploit RNA for diagnostic and therapeutic purposes. The various research lines converge on studying the role of non-coding RNA in cancer and on the utility of extracellular RNA in liquid biopsies. Through a combination of high-throughput (functional) genomics technologies and bio-informatics tools, we seek to answer various fundamental and translational research questions.
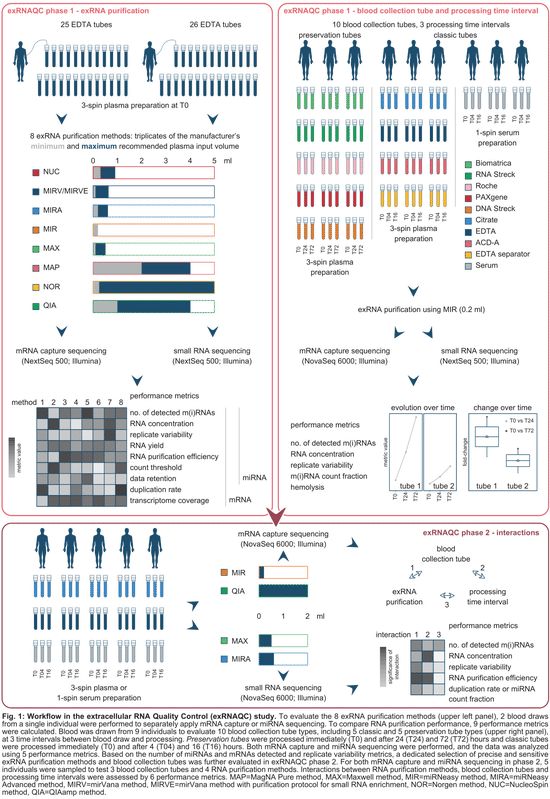
controlling pre-analytical variables of RNA sequencing in blood-based precision medicine
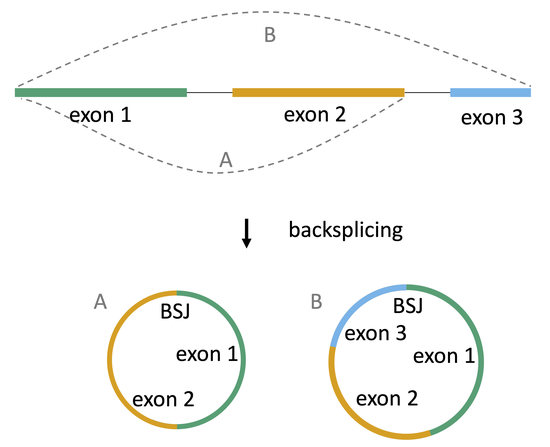
circular RNAs in cancer
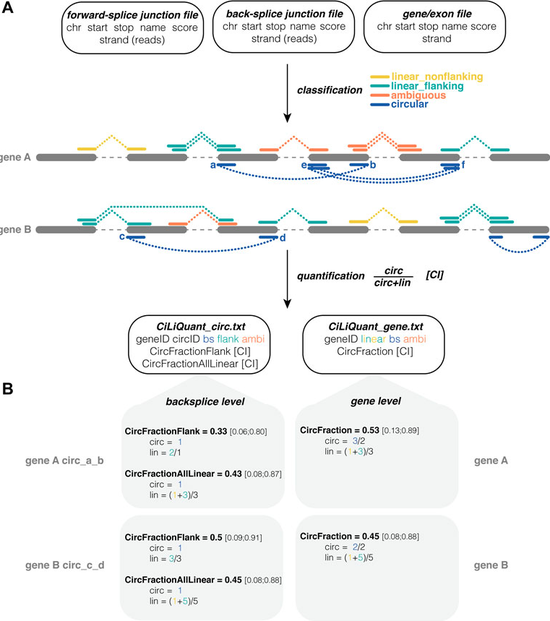
tool to determine the relative circular and linear abundance of transcripts
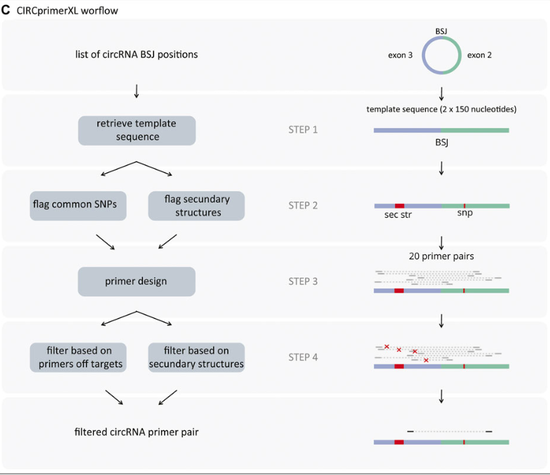
tool for primer design for circRNAs
digital PCR data-analysis tools developed by the Ghent University Digital PCR Consortium
crowdsourcing knowledgebase with experimental parameters of public extracellular vesicles (EV) studies

antisense oligonucleotide design for long non-coding RNAs
a comprehensive compendium of human long non-coding RNAs

easy-to-use online database that keeps track of all historical and current miRNA annotation present in the miRBase database
microRNA target prediction
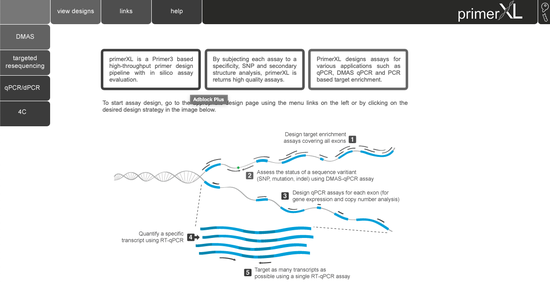
state-of-the-art PCR assay design for quantitative and digital PCR, targeted resequencing, double-mismatch allele specific PCR (DMAS), and 4C

public database for sharing of quantitative and digital PCR using the RDML data exchange format
a custom paltform for spatial transcriptomics

visualization and analysis method to evaluate human transcription start sites in relation to publicly available CAGE-sequencing, ChIP-sequencing and DNase-sequencing datasets

Non-coding RNA, Extracellular RNA, Liquid biopsies, Pan-cancer, Method development
Non-coding RNA, Cancer, Extracellular RNA, Liquid biopsies, Bioinformatics, Immune oncology
Cancer, Extracellular RNA, Liquid biopsies, Pediatric tumors, Cancer diagnostics, detection and prognosis
Spatial Transcriptomics, Resistance, Neuroblastoma, Pleiotropic treatment effects
Cancer, Non-coding RNA, Extracellular RNA, Liquid biopsies, Pediatric tumors, Spatial transcriptomics

Cancer, Bioinformatics, Genetics, Lab-test, Singlet oxygen photo-electrochemical detection
Photo-electrochemistry, Singlet oxygen, Cancer biomarkers, Nucleic acids, Diagnostics




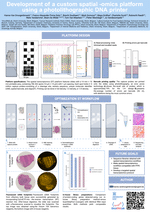




Accurate assessment of treatment response and residual disease is indispensable for the evaluation of cancer treatment efficacy. However, performing tissue biopsies for longitudinal follow-up poses a major challenge in the management of solid tumours like neuroblastoma. In the present study, we evaluated whether circulating miRNAs are suitable to monitor neuroblastoma tumour burden and whether treatment-induced changes of miRNA abundance in the tumour are detectable in serum. We performed small RNA sequencing on longitudinally collected serum samples from mice carrying orthotopic neuroblastoma xenografts that were exposed to treatment with idasanutlin or temsirolimus. We identified 57 serum miRNAs to be differentially expressed upon xenograft tumour manifestation, out of which 21 were also found specifically expressed in the serum of human high-risk neuroblastoma patients. The murine serum levels of these 57 miRNAs correlated with tumour tissue expression and tumour volume, suggesting potential utility for monitoring tumour burden. In addition, we describe serum miRNAs that dynamically respond to p53 activation following treatment of engrafted mice with idasanutlin. We identified idasanutlin-induced serum miRNA expression changes upon one day and 11 days of treatment. By limiting to miRNAs with a tumour-related induction, we put forward hsa-miR-34a-5p as a potential pharmacodynamic biomarker of p53 activation in serum.

Blood plasma, one of the most studied liquid biopsies, contains various molecules that have biomarker potential for cancer detection, including cell-free DNA (cfDNA) and cell-free RNA (cfRNA). As the vast majority of cell-free nucleic acids in circulation are non-cancerous, a laboratory workflow with a high detection sensitivity of tumor-derived nucleic acids is a prerequisite for precision oncology. One way to meet this requirement is by the combined analysis of cfDNA and cfRNA from the same liquid biopsy sample. So far, no study has systematically compared the performance of cfDNA and cfRNA co-purification to increase sensitivity. First, we set up a framework using digital PCR (dPCR) technology to quantify cfDNA and cfRNA from human blood plasma in order to compare cfDNA/cfRNA co-purification kit performance. To that end, we optimized two dPCR duplex assays, designed to quantify both cfDNA and cfRNA with the same assays, by ensuring that primers and probes are located within a highly abundant exon. Next, we applied our optimized workflow to evaluate the co-purification performance of two manual and two semi-automated methods over a range of plasma input volumes (0.06–4mL). Some kits result in higher nucleic acid concentrations in the eluate, while consuming only half of the plasma volume. The combined nucleic acid quantification systematically results in higher nucleic acid concentrations as compared to a parallel quantification of cfDNA and cfRNA in the eluate. We provide a framework to evaluate the performance of cfDNA/cfRNA co-purification kits and have tested two manual and two semi-automated co-purification kits in function of the available plasma input amount and the intended use of the nucleic acid eluate. We demonstrate that the combined quantification of cfDNA and cfRNA has a benefit compared to separate quantification. We foresee that the results of this study are instrumental for clinical applications to help increase mutation detection sensitivity, allowing improved disease detection and monitoring.

While cell-free DNA (cfDNA) is widely being investigated, free circulating RNA (extracellular RNA, exRNA) has the potential to improve cancer therapy response monitoring and detection due to its dynamic nature. However, it remains unclear in which blood subcompartment tumour-derived exRNAs primarily reside. We developed a host-xenograft deconvolution framework, exRNAxeno, with mapping strategies to either a combined human-mouse reference genome or both species genomes in parallel, applicable to exRNA sequencing data from liquid biopsies of human xenograft mouse models. The tool enables to distinguish (human) tumoural RNA from (murine) host RNA, to specifically analyse tumour-derived exRNA. We applied the combined pipeline to total exRNA sequencing data from 95 blood-derived liquid biopsy samples from 30 mice, xenografted with 11 different tumours. Tumoural exRNA concentrations are not determined by plasma platelet levels, while host exRNA concentrations increase with platelet content. Furthermore, a large variability in exRNA abundance and transcript content across individual mice is observed. The tumoural gene detectability in plasma is largely correlated with the RNA expression levels in the tumour tissue or cell line. These findings unravel new aspects of tumour-derived exRNA biology in xenograft models and open new avenues to further investigate the role of exRNA in cancer.
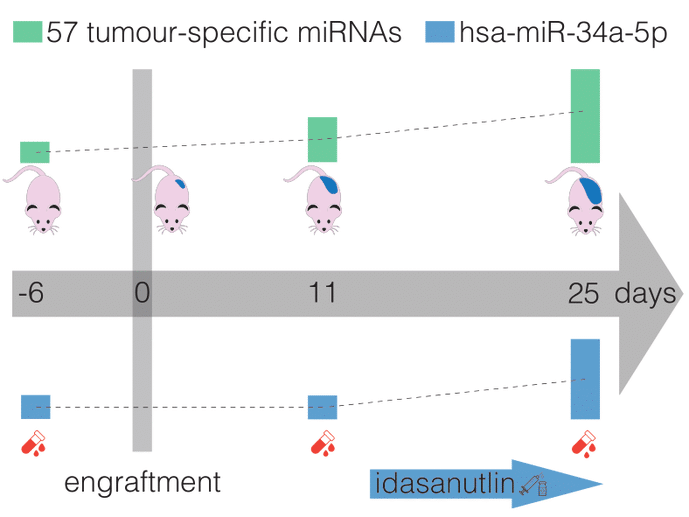
We assess the performance of mRNA capture sequencing to identify fusion transcripts in FFPE tissue of different sarcoma types, followed by RT-qPCR confirmation. To validate our workflow, six positive control tumors with a specific chromosomal rearrangement were analyzed using the TruSight RNA Pan-Cancer Panel. Fusion transcript calling by FusionCatcher confirmed these aberrations and enabled the identification of both fusion gene partners and breakpoints. Next, whole-transcriptome TruSeq RNA Exome sequencing was applied to 17 fusion gene-negative alveolar rhabdomyosarcoma (ARMS) or undifferentiated round cell sarcoma (URCS) tumors, for whom fluorescence in situ hybridization (FISH) did not identify the classical pathognomonic rearrangements. For six patients, a pathognomonic fusion transcript was readily detected, i.e., PAX3-FOXO1 in two ARMS patients, and EWSR1-FLI1, EWSR1-ERG, or EWSR1-NFATC2 in four URCS patients. For the 11 remaining patients, 11 newly identified fusion transcripts were confirmed by RT-qPCR, including COPS3-TOM1L2, NCOA1-DTNB, WWTR1-LINC01986, PLAA-MOB3B, AP1B1-CHEK2, and BRD4-LEUTX fusion transcripts in ARMS patients. Additionally, recurrently detected secondary fusion transcripts in patients diagnosed with EWSR1-NFATC2-positive sarcoma were confirmed (COPS4-TBC1D9, PICALM-SYTL2, SMG6-VPS53, and UBE2F-ALS2). In conclusion, this study shows that mRNA capture sequencing enhances the detection rate of pathognomonic fusions and enables the identification of novel and secondary fusion transcripts in sarcomas.
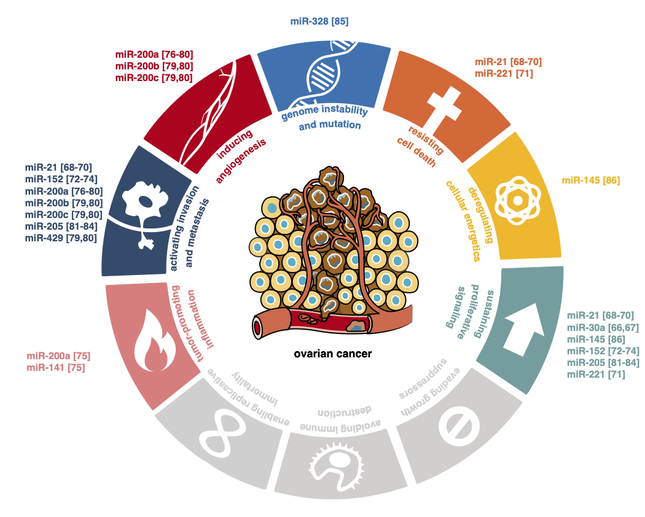
Ovarian cancer is often diagnosed in an advanced stage and is associated with a high mortality rate. It is assumed that early detection of ovarian cancer could improve patient outcomes. Unfortunately, effective screening methods for early diagnosis of ovarian cancer are still lacking. Extracellular RNAs circulating in human biofluids can reliably be measured and are emerging as potential biomarkers in cancer. In this systematic review, we present 75 RNA biomarkers detectable in human biofluids that have been studied for early diagnosis of ovarian cancer. The majority of these markers are microRNAs identified using RT-qPCR or microarrays in blood-based fluids. A handful of studies used RNA-sequencing and explored alternative fluids, such as urine and ascites. Candidate RNA biomarkers that were more abundant in biofluids of ovarian cancer patients compared to controls in at least two independent studies include miR-21, the miR-200 family, miR-205, miR-10a and miR-346. Amongst the markers confirmed to be lower in at least two studies are miR-122, miR-193a, miR-223, miR-126 and miR-106b. While these biomarkers show promising diagnostic potential, further validation is required before implementation in routine clinical care. Challenges related to biomarker validation and reflections on future perspectives to accelerate progress in this field are discussed.
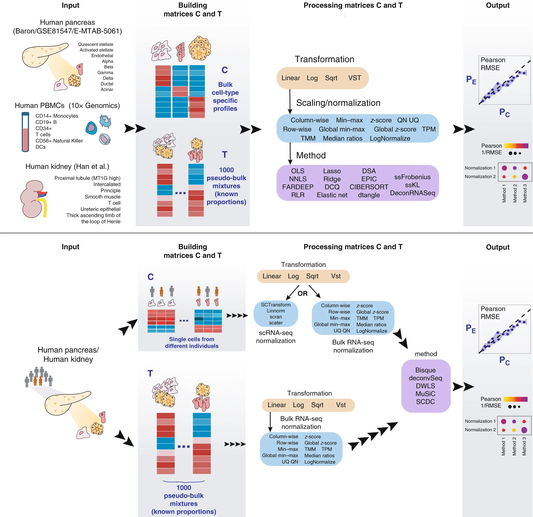
Many computational methods have been developed to infer cell type proportions from bulk transcriptomics data. However, an evaluation of the impact of data transformation, pre-processing, marker selection, cell type composition and choice of methodology on the deconvolution results is still lacking. Using five single-cell RNA-sequencing (scRNA-seq) datasets, we generate pseudo-bulk mixtures to evaluate the combined impact of these factors. Both bulk deconvolution methodologies and those that use scRNA-seq data as reference perform best when applied to data in linear scale and the choice of normalization has a dramatic impact on some, but not all methods. Overall, methods that use scRNA-seq data have comparable performance to the best performing bulk methods whereas semi-supervised approaches show higher error values. Moreover, failure to include cell types in the reference that are present in a mixture leads to substantially worse results, regardless of the previous choices. Altogether, we evaluate the combined impact of factors affecting the deconvolution task across different datasets and propose general guidelines to maximize its performance.
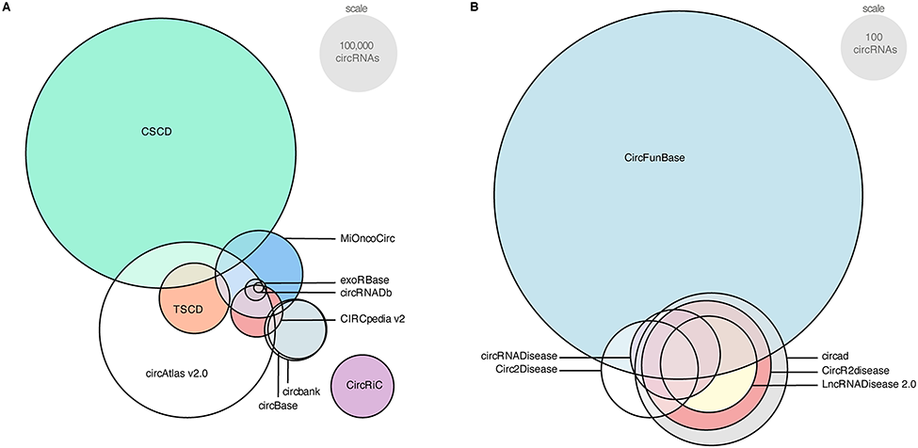
Circular RNAs (circRNAs) are covalently closed RNA molecules that have been linked to various diseases, including cancer. However, a precise function and working mechanism are lacking for the larger majority. Following many different experimental and computational approaches to identify circRNAs, multiple circRNA databases were developed as well. Unfortunately, there are several major issues with the current circRNA databases, which substantially hamper progression in the field. First, as the overlap in content is limited, a true reference set of circRNAs is lacking. This results from the low abundance and highly specific expression of circRNAs, and varying sequencing methods, data-analysis pipelines, and circRNA detection tools. A second major issue is the use of ambiguous nomenclature. Thus, redundant or even conflicting names for circRNAs across different databases contribute to the reproducibility crisis. Third, circRNA databases, in essence, rely on the position of the circRNA back-splice junction, whereas alternative splicing could result in circRNAs with different length and sequence. To uniquely identify a circRNA molecule, the full circular sequence is required. Fourth, circRNA databases annotate circRNAs’ microRNA binding and protein-coding potential, but these annotations are generally based on presumed circRNA sequences. Finally, several databases are not regularly updated, contain incomplete data or suffer from connectivity issues. In this review, we present a comprehensive overview of the current circRNA databases and their content, features, and usability. In addition to discussing the current issues regarding circRNA databases, we come with important suggestions to streamline further research in this growing field.
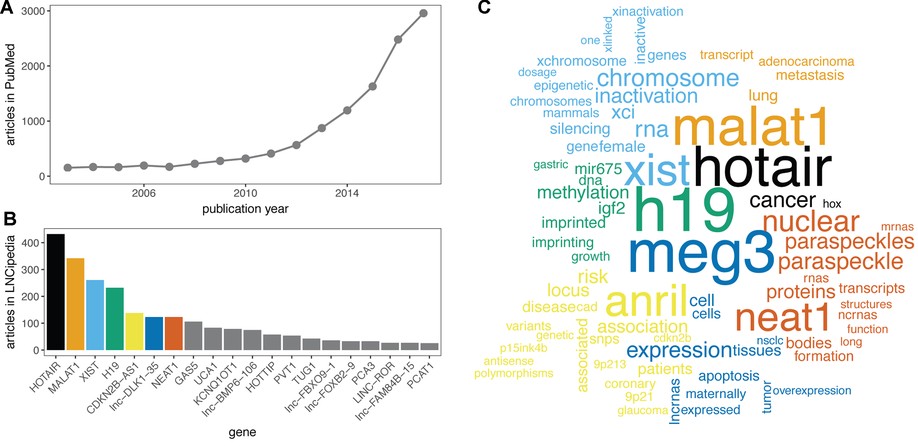
Here, we present the fifth release of the human lncRNA database LNCipedia ( https://lncipedia.org). The most notable improvements include manual literature curation of 2482 lncRNA articles and the use of official gene symbols when available. In addition, an improved filtering pipeline results in a higher quality reference lncRNA gene set.